 扫一扫,手机浏览
扫一扫,手机浏览- 技术文章
解锁 OptiMelt 熔点仪:材料、制药、化工、橡胶、化妆品纯度检测密码
2025-02-08 11:39:20 来源:北京卓立汉光仪器有限公司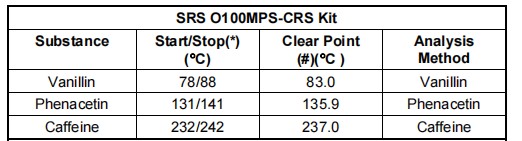
背景:
MP (Melt Piont)熔点的定义是物质从固态变为液态的温度;在纯结晶物质熔化过程中,注入此物质的所有能量都以熔化热的形式消耗掉,此时物质本体温度保持恒定,直至所有物质由固相融化为液相(即完成熔化过程)。物质熔化是一个高度可控过程,物质熔点是一个可以高精度标定的参数。
纯结晶物质的熔点清晰且明确。我们可以在化学*业网站或官*药典内方便的查询到。
对于物质的熔点检测与分析是检验物质纯度*古老的鉴定和测试方法之一,因为少量的杂质也会改变熔点,或至少明显地扩大其熔点范围。
熔点检测仪的样品制备与仪器操作简便,设备价格与耗材成本低廉,测试结果准确且重复性高,这诸多优势使其广泛应用于:
・ 制药行业
・ 化学与化工
・ 橡胶与轮胎
・ 等等
美国SRS公司OptiMelt全自动熔点仪
美国SRS公司OptiMelt全自动熔点仪以其测试结果准确,稳定,使用方便且用户界面友好等优点为广大用户所喜爱,今天就从OptiMelt用户界面友好以及操作技巧两个方面做一个小小的总结:

图1. 美国SRS公司OptiMelt全自动熔点仪
一.OptiMelt以用户界面友好的全自动检测。
・ 用户只需在触摸屏上选择起始温度,温度梯度,终止温度等参数后,按下开始键,OptiMelt即可自动开始测试,并利用内置的高清数码相机采集实时图像,并通过内置程序自动检测熔融状态,
・ 用户还可同时利用带放大镜的目视窗口观察当前熔融状态,并可随时按触摸屏增加多至6个的个性标注点
・ 图形化软件界面,所有带时间轴与标注点的图像可后期调用,以便检验与备查。
・ 内置未用毛细管和已用毛细管储槽(如图)避免混杂。
・ 到达起始温度声光提示插入毛细管,到达终止温度声光提示终了状态
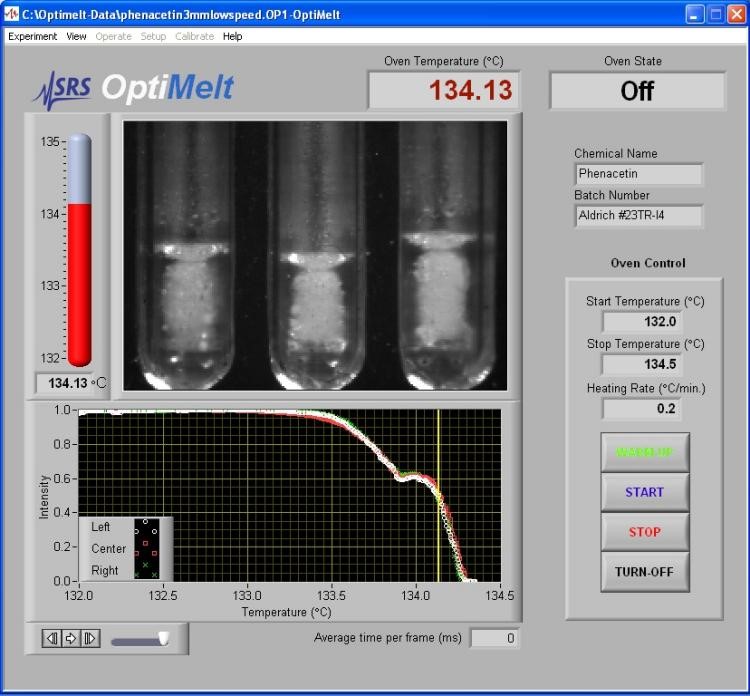
图2. 美国SRS公司OptiMelt全自动熔点仪软件操作界面
二.如何利用OptiMelt自动化熔点检测仪准确的检测熔点
1.样品制备:
任何被装入熔点毛细管的物质必须:
1)完*干燥
2)均匀
3)粉末状
潮湿的样品建议先干燥,比如在有P2O5(五氧化二磷)干燥剂的干燥皿中静置48小时。
优质样品呈细粉末形式,好处一是可以使得热传导均匀且迅速,好处二是具备更高更均匀的反射率便于设备相机准确辨别溶解状态。所以粗晶和非均质样品必须在研钵中粉碎成细粉末。建议使用玛瑙、玻璃或氧化铝研钵和杵。

图3. 干燥的样品也需要精细的研磨
要将粉末样品装入毛细管,只需从毛细管开口的一端轻轻压入样品几次。然后翻转毛细管在坚硬表面上轻敲几次(首*方法),使得样品自动落在毛细管底部。或者,可使用设备自带的细铁丝(通条)插入毛细管进一步压实样品,提高测量的重复性。

图4. 将样品压入毛细管,轻敲底部,或者用通条压实
为了获得*佳测量结果和重复性,建议样品高度在2.0mm至3.0mm之间,样品高度过高会由于样品量过多而需要额外的加热量,过低会由于样品量不足反而加大熔化温度变化范围。
如果您的样品是吸湿性的,或会在高温下升华,在填充样品后毛细管的开口端建议熔融密封。吸湿性样品在两次试验之间必须存放在干燥皿中。这点在潮湿的环境中或雨天尤为重要。
2.毛细管的使用注意事项

图5. 毛细管插入管槽后会自由滑落到底部
将包含样品的毛细管插入SRS公司OptiMelt熔点测试仪顶部的样品加热槽中。加热槽*多可同时容纳三根毛细管。由于每根毛细管都是一个热载荷,为了提高测试结果的重复性,即使某毛细管没有填充样品,也建议插满三根管。或者三根毛细管中填充相同的物质并取其熔点温度平均值,这是提高熔点检测准确度与重复性*快捷和*简单的方法。
提示:
通常认为,在将毛细管插入加热架之前,用干净的布擦拭其外表面是一种良好的做法。随着时间的推移,灰尘会积聚在加热块的玻璃观察窗上,降低熔融的整体能见度,所以也建议定期擦拭。
用通条过于紧密压实毛细管内样品也不太好,往往会导致在熔化过程中形成过多的气泡,从而影响对弯月面和澄清点的正确检测。
大多数药典清单推荐了熔点样品的干燥程序和经认证的参考标准,请注意查询与参考。
在将任何毛细管放入样品槽之前,确保OptiMelt熔点分析仪已接电和设置初始温度低于样品预期熔点温度。
使用同一批毛细管进行校准和常规测量,以确保结果的重复性。不是所有的毛细血管能保持良好的一致性,请选用可信赖的商品。

图6. 商品毛细管
标准的OptiMelt熔点分析仪包括一小罐专门设计的毛细管:(1)适合OptiMelt熔点分析仪加热支架,(2)提供*均匀和可再现的结果。替换毛细管可以直接从SRS或SRS官网指*代理商购买。
千万不要将毛细管强行放入加热槽中!一旦毛细血管插入样品孔中,它就会自行滑落到支架的底部。
有些化学家更喜欢自己制造的毛细管。为了获得准确和可重复的结果,一般不建议自制。强烈建议使用严格控制制造公差的商品毛细管。
为了精确测量,建议严格遵守2mm至3mm的*佳填充高度。
在可能的情况下,使用通条将毛细管底部的样品轻轻压实。从SRS购买的每包毛细管都包含一根通条。
如果你必须自己制作毛细管,请确保用含中性洗涤剂的稀释溶液清洗玻璃管内部,然后用稀释(10%)盐酸冲洗,*后用蒸馏水彻*冲洗。因为毛细管内表面残留的碱性物质是导致低熔点和宽熔化范围的主要原因之一。而采购可信赖的商品毛细管一般不需要考虑预清洗环节。
3. 仪器设置:

图7. 小型铝制加热炉
除了适当的样品制备外,仔细选择仪器设置对于准确和可再现性的熔点测定也是必*可少的。
熔点仪器的现代趋势是小型铝制加热炉。小型加热炉*大限度地减少了过冲,这使得操作者可以将初始温度设置得更接近熔化温度,从而减少了分析时间。一个典型的加热炉可以容纳三根毛细管,三根管子周围的热质量非常接近。在熔化过程中,三根毛细管之间的偏差通常低至0.02C到0.1C(取决于设定温度)。
小型加热炉的主要优点是没有过冲。这使您可以将起始温度设置在低于化合物预期熔点的<5°C,其可以快速加热和冷却,并允许仅持续2至3分钟的测定。
几乎每一种现代熔点分析仪器都遵循药典标准熔点测定程序,包括四个基本步骤:
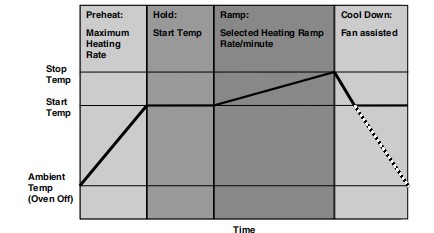
图8. 一个完整的测试流程
步骤一、加热炉迅速预热到用户指*的起始温度,选择起始温度仅比样品的预期熔点低几摄氏度。
步骤二、一旦温度稳定,将多达三个样品毛细血管插入加热炉中,在温度稳定后,立即启动加热斜坡。
步骤三、样品的温度继续以用户指*的升温速率上升,直到达到用户指*的停止温度。
在此期间,自动或同时目视观察的熔点、熔化温度范围和其他热相关过程被记录下来。
步骤四、熔化过程完毕后,建议将毛细管与样品一并丢弃,不再考虑清洗并重复利用,加热炉迅速冷却回起始温度,为新的测定做准备。
正确选择起始温度、加热斜坡和停止温度是绝对必要的,以防止由于不正确或过快的样品加热而导致的结果不准确。
1) 起始温度(OptiMelt熔点分析仪范围:30℃-400℃)
这是样品毛细管被放置加热架时的温度,并作为加热斜坡的起始温度。起始温度通常设定在物质的预期熔点温度以下5°C至10°C。
提示:
起始温度必须至少高于环境温度10°C,以确保稳定。
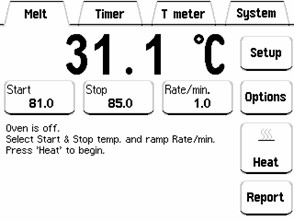
图9. 在面板上设置起始,终了温度,加热斜坡速率
2)加热斜坡速率
(OptiMelt熔点分析仪范围:0.1-20℃/分钟)
这是加热斜坡开始温度和*终温度之间的固定温升速率。用户可调斜坡率是现代自动化熔点仪器的*备功能。
加热斜坡是影响熔点测定精度的*重要的仪器参数。
由于熔点温度不是在物质内部直接测量的,而是在毛细管外部(即在加热炉内部)测量的,我们知道,在样品完*熔化之前,纯熔融物质的温度保持恒定。然而,在这段时间内,加热炉仍然在根据您所选择的加热速率继续升温(即热滞后)。也就是说仪器当前显示的温度并不真正对应于熔化物质的确切熔点温度,而是对应于当前加热炉的温度。因此,加热炉温度上升得越快,测得的熔点与真实熔点之间的差值就越大。就此问题,一些熔点检测仪(如SRS 公司OptiMelt)可以依据药典做温度读数的校正与补偿,以便可以报告纯物质的“真正热力学”熔点。
注意:
误用过快的加热斜坡速率是熔点测量不准确的主要原因。
对于常规测定,加热速率建议2°C/min或以下。较高的速率只推荐用于熔点未知的物质的快速测定。做样品纯度测定和熔点精确测量建议设置在≈0.5°C/min或以下,如果可接受较长实验时间,可以考虑保持在0.1至0.2°C/min.的加热斜坡速率。还有某些特殊情况,比如样品在低于物理熔点的温度下已经开始化学分解了,此时通常设置高于5°C/min的高加热斜坡速率,以尽量规避样品分解副产物带来的污染。混合熔点测定甚至可以在加热斜坡高达10°C/min的情况下进行。
提示:
根据大多数药典建议,熔点记录中应始终包括:加热速率、熔点范围,以便得到可重复的准确结果。
进行初步(即快速)熔点测定,迅速升温(10到20°C/min),通常可以节省时间。在获得近似熔点后,以小得多的加热斜坡进行第二次测定,起始温度比预期熔点低5°C。第二次测定必须使用新鲜样品。
终止温度
(OptiMelt熔点分析仪范围: [起始温度+5°C] - 400°C)
这是加热斜坡终止时的温度。在达到终止温度后,请将样品加毛细管丢弃,而测试结果已经保存并显示在测试报告中,加热炉会自动冷却回起始温度,为新一轮的测定做准备。
注意:旧样品不要重新熔化!!总是用洁净的毛细管开始新的实验。
4.仪器校准:
OptiMelt熔点分析仪建议每6个月校准一次。
校准程序非常简单。测量熔点在75、125和225℃左右的三种认证参考标准物质CRSs的熔点,然后与它们的认证值进行比较。如果两组数字(测量值与认证值)相互偏离超出了仪器的精度,则必须重新校准加热块中的温度测量值。这一切可通过OptiMelt前面板的操作界面按步骤完成。
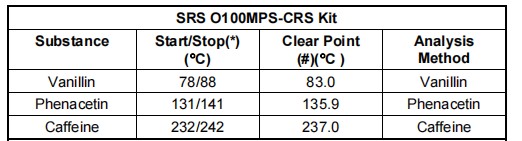
符合认证参考标准的CRS试剂盒(SRS公司产品编码 Part# O100MPS)可追溯至世卫组织国际药典标准,可直接从SRS斯坦福研究系统或其代理商处购得,包含如下标定物质:
4.视觉观察:
在熔点测定过程中,毛细血管内的样品会发生一些明显的变化。在解释升温过程中发生物理和化学变化时的主观视觉观察可能是影响熔点结果测试可重复性的一个重要因素。
观察者应注意以下事件,并记录它们的温度,以完善在熔化过程中观察到的样品变化的完整记录。
*初的变化迹象
记录样品中*初的变化迹象。早期的变化是由于:
(1)溶剂损失(如果样品未被良好干燥处理而在加热时发生的脱水)
(2)结晶状态变化(萎*)
(3)开始缓慢分解(变暗或变色)
(4)溶剂在管内*冷点开始冷凝
(5)单个孤立的晶体开始熔化,刚刚开始出现某些孤立的液滴,即Sintering Point熔结点
起熔点OnSet Point
起熔点OnSet Point通常被认为是熔化的“正式”开始;液体首*清晰地作为与晶体共存的独立相出现。它不能与“熔结点Sintering Point”相混淆,“熔结点Sintering Point”对应的是由于一些晶体表面熔化而产生的某些孤立液滴。
提示:
起熔点对应于物质的熔点范围的低温端被记录。
美国药典将起熔点描述为“被测物质柱被观察到对着试管管壁发生坍塌时的温度”。这被称为样本的崩溃
对于依赖于测试样品整体吸收率或反射率变化的自动化仪器,美国药典将起熔点描述为“在熔化过程中观察到*一个检测信号发生变化的温度”。
依赖于测试样品整体吸收率或反射率变化的自动化仪器不能很容易地探测到熔化的起熔点。它们通常报告的熔化起始点温度比肉眼检测到的温度要高一点。这是因为仪器检测到整体吸收率或反射率变化时,样品的外观在此之前就已经发生显著变化。所以检测整体吸收率或反射率变化确定起熔点会导致熔化温度范围的缩小,这在一些分析和品控应用中可能是一个值得关注的问题。
OptiMelt熔点分析仪可以自动检测并记录样品的起熔点。内置摄像头对样品物理外观的微小变化都很敏感,与肉眼的灵敏度非常接近。用户可调节阈值(起熔点%)可用于仔细匹配每种物质的起熔点的目视值和自动测量值。
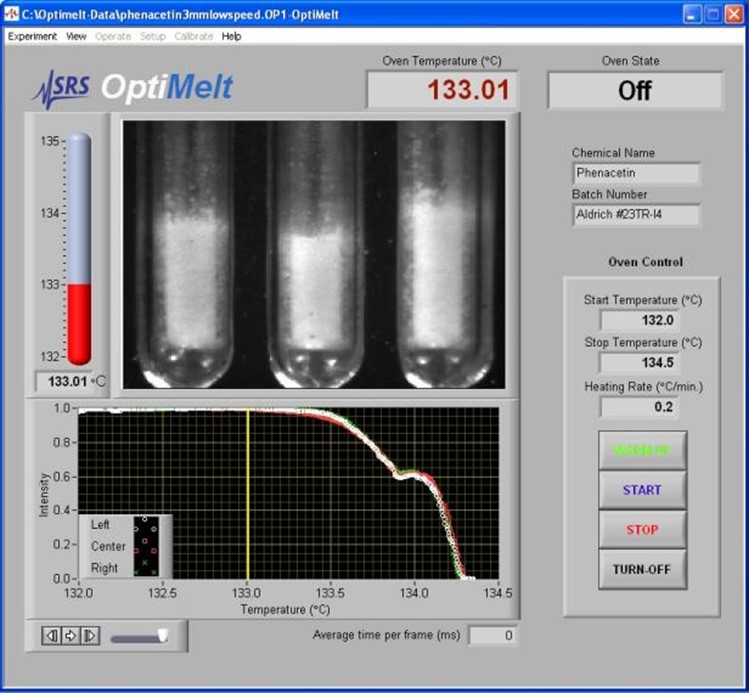
图10. 操作软件显示的OnSet 起熔点
弯月面点Meniscus Point
弯月面点Meniscus Point对应于样品熔化中的阶段,此时液体的弯月面清晰可见;中下部是固相,顶部是清晰的液相,有清晰可见的弯月面。这一点很容易检测到,除非偶尔有气泡从下面把未熔化的固体推到表面。
提示:
弯月面点通常是欧洲熔点表中列出的熔点温度和英国药典方*论的首*值。
弯月面点是政府化验师实验室(LGC)为每项熔点标准所记录的三个数值(起熔点、弯月面点及澄清点)之一。为了消除检测中的主观性,LGC将弯月面点定义为“可以看到明确的弯月面,并且毛细管中有等量的固体和液体的点”。
美国药典熔点方法(Method <741> of USP25-NF20)没有特别提到弯月面点。澄清点被认定为一种物质的“熔点”。请注意,这是英国和美国药典在熔点解释上的巨大差异。
SRS OptiMelt熔点分析仪可以自动检测和记录样品的弯月面点。用户可调阈值(Single %)可匹配弯液面点的视觉和自动记录。
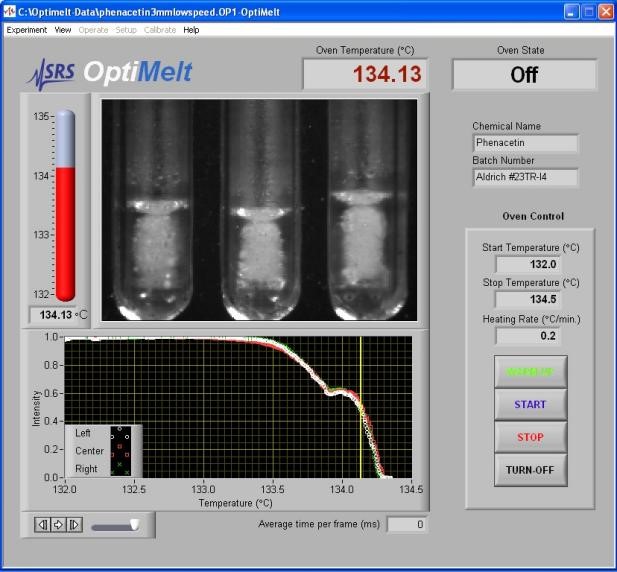
图11. 操作软件显示的弯月面点Meniscus Point
澄清(或液化)点 Clear Point
澄清(或液化)点 Clear Point对应于完*熔化的阶段,在这个阶段物质完*变成液体,没有固体留下(即*后的晶体完成熔化)。
澄清点比起始点更被加热斜坡速率严重影响。一般来说,澄清点随着斜坡率的增加而增加(见表1)。
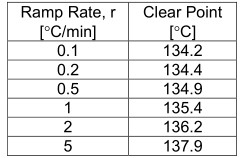
表1.非那西丁在不同斜坡速率下的澄清点(OptiMelt系统)。
提示:
澄清点记录对应于记录物质熔点范围的高温端。
澄清点通常是熔点表中列出的单一熔点温度。
澄清点是在美国药典的熔点表中*常见的温度,也是美国药典唯*接受的作为物质“单一”熔点的温度。
依靠于整体光学吸收和反射的简单自动化检测系统能够给出与澄清点*相关数字。
SRS OptiMelt熔点分析仪可以自动检测和记录样品的澄清点。用户可调整阈值(Clear %),以匹配澄清点的视觉记录和自动记录。
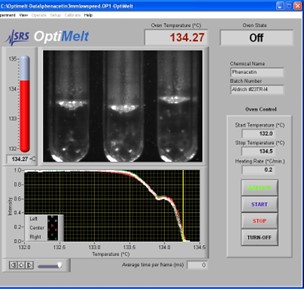
图12. 操作软件显示澄清(或液化)点 Clear Point
样品变化的其他迹象
在澄清点之前、期间和之后,样品的任何变化也可以被记录下来。常见迹象包括:
升华:晶体重新结晶在毛细管的上部管壁上。
分解:样品在熔化过程中和熔化后出现气泡或颜色变化。
SRS OptiMelt熔点分析仪的用户可以利用肉眼观察图像,并随时点击仪器前面板的左,中,右按钮记录对应样品的个性化记录点。
图13. 手工点击面板按钮可增加个性化记录点
熔点范围Melting Point Range
在动态熔点测量中,永远不会实现固液相之间的真正平衡,熔点范围一般被定义为起熔点和澄清点之间的间隔,是一个有价值的固体化合物纯度指标。
熔点范围是在科学论文、标准检测流程、参考表和熔点标准中*常见的熔点记录项目。记录一种物质的整个熔点范围总是有益处的,特别是对于(1)未知的或新的化合物,(2)不纯的样品,(3)熔点间隔较大的混合物和(4)多形化合物。观察到的熔点范围有助于识别该物质,并得出有关其纯度和热稳定性的结论。
记录固体样品的熔化范围[起熔点,澄清点],以及斜坡速率,是记录熔点测试结果的首*方法,它比单一数字记录要可靠得多。
如果必须使用单一数字记录,请指*是使用澄清点还是弯液面点。
斜坡速率影响熔点范围记录,其设置必须始终完*符合GLP规范。
表2.非那西丁在不同斜坡速率下的熔点范围(SRS OptiMelt熔点分析仪)。
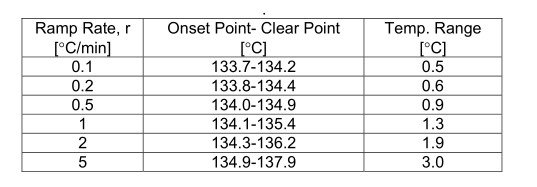
如表格所示,斜坡速率在对于澄清点上的影响远对起始点上的影响更大。
提示:
SRS OptiMelt熔点分析仪可以自动检测和记录样品的起熔点和澄清点。用户可调阈值(起始%,透明度%)可用于匹配两个点的视觉值和自动值。
绝大多数纯有机化合物在1.5℃的温度范围内熔化,或在加热速率为0.5C/min以下的情况下,在较小的温度范围内(≈2℃)熔化。而一些有机混合物(比如氨基酸、酸盐、胺盐、碳水化合物等)会在大的多的温度范围内熔化。
不纯的物质(即混合物)会在或大或小的温度范围内熔化。
如何出具测试报告
一份完整的熔点报告应包括足够的信息,以便其他人能够重现测定结果并比较结果。Carter and Carter (J. Chem. Ed., 72 (1995) 647) 提出了非常有用的报告指南,与现代GLP规范要求兼容。现转载如下:
记录所有仪器设置,特别是加热速率,以便重现实验结果或合理地作出调整。
对于常规熔点范围,起熔点和澄清点温度差尽量接近于0.1°C(或*多为0.5°C)。
对于重要的熔点范围,如新化合物的熔点范围,报告记录起熔点、弯月面点和澄清点应尽量接近于0.1°C。
如果要在报告中以单一温度作为熔点(不推荐),请标明是使用弯月面点还是澄清点。
使用众所*知的熔点标准品(即CRS认证参考标准)进行设备校准。
熔点参考表
对于熔点表中关于究竟需要列出什么信息通常存在一些不确定性,特别是在列出物质熔点的单一温度时。
这种混淆是因为:虽然大多数化学家使用澄清点来记录样品的熔点温度,但其他人更喜欢用弯月面点来记录。弯月面点通常被认为更接近真实的热力学值,因为它对应于毛细管中液体和固体共存的状态。然而,这一假设并没有真正的热力学依据。幸运的是,在大多数情况下,两个数字之间(即澄清点与弯月面点)的差异非常小,并且在大多数测定的公差范围内。
熔点降低/熔化温度范围
在液相状态不相溶的混合物,会有熔点降低的现象,且会呈现一个熔化的温度范围(或间隔),而不是一个明确且尖锐的熔点。
熔点降低的范围取决于混合物的组成。熔点的降低可用于测定化合物的纯度和特性。
经验法则
1%的外来杂质会导致熔点降低约0.5°C。
这就是为什么记录熔点范围是熔点测定的首*报告格式,也是比单一温度熔点记录更有用的主要原因。
熔点范围较大通常表明被测试物质是不纯的,但也可能是由于纯物质在达到相变之前经历了某些分解,在加热过程中分解的纯物质的副产物,形成母体物质和副产物的混合物,往往会显著增大熔化温度范围。在某些情况下,材料在低于真正熔点的温度下会经历轻微的液化和收缩。在其他情况下,材料可能会严重分解和变色,以至于无法观察到准确的熔点。
纯度跟踪
熔点降低现象可应用于合成化合物纯度的评定。
在制备有机化学物质过程中,物质的纯度通常必须在尚无纯净的参考样品的情况下进行评定。例如,在尝试制造一种新的化合物时,就是这样的情况。原料通常要经过几个再结晶步骤,并在每个阶段确定熔点。起熔点持续增加,熔化温度范围持续减小,直到物质变得纯净,或者直到它达到通常所采用的纯化方法所能达到的纯度。
提示:
通常的做法是将反应的合成产物重新结晶,直到在其熔点范围内检测不到更多的变化。
混合熔点
如果两种样品在相同的温度下熔化,混合熔点测定可以揭示它们是否是同一种物质。
熔点降低现象可应用于鉴定未知纯度的物质。例如,如果您测量样品熔点在160°C,您检索药典的熔点表会发现,这代表了几种不同的参考化合物,但是它们的熔点相同。该物质可以利用它的混合熔点来确定:样品与少量的参考样品逐个混合,并确定每种情况下的混合熔点。当样品被混入少量的参照物而熔点降低时,这两种物质不可能是相同的。然而,如果混合物的熔点不下降,则加入的参照物与样品相同(即样品已被鉴定)。
混合熔点技术是所有高质量熔点分析仪器在其加热槽中至少可容纳三个毛细管的重要原因。
*常见的混合熔点测量方法是,要确定三个熔点:(1)样品,(2)参考物,(3)参考物和样品以1:1的比例混合。如果混合物的熔点保持不变,则这两种物质是相同的。如果熔点降低,它们就是两种不同的物质。
提示:
混合熔点检测对精度和可重复性的要求不像做高精度、单熔点测定时那么高。加热速率高达10°C/min可用于混合熔点测定,这样可以大幅度节省实验用时。
有几对物质在混合时熔点并不下降,但更常见的是,只有在某些配比下才观察到这种熔点不下降的情况。规避这种问题只需要一点额外的工作,通常是制备20/80,50/50,80/20%的样品/参考物三种比例的混合物并在熔点分析仪中使用三根毛细管同时进行熔点测量。如果这三种混合物在相同的温度下熔化,那么这两种化合物很可能是同一种化合物!
-
 北京卓立汉光仪器有限公司联系电话
北京卓立汉光仪器有限公司联系电话13810146393
内容声明:谷瀑为第三方平台及互联网信息服务提供者,谷瀑(含网站、客户端等)所展示的商品/服务的标题、价格、详情等信息内容系由店铺经营者发布,其真实性、准确性和合法性均由店铺经营者负责。谷瀑提醒您购买商品/服务前注意谨慎核实,如您对商品/服务的标题、价格、详情等任何信息有任何疑问的,请在购买前通过谷瀑与店铺经营者沟通确认;谷瀑上存在海量店铺,如您发现店铺内有任何违法/侵权信息,请在谷瀑首页底栏投诉通道进行投诉。北京卓立汉光仪器有限公司 电话:010-56370168-696 手机:13810146393 地址: 北京市中关村科技园区通州园金桥产业基地环科中路16号,联东U谷中试区68号B座











